结构变异(Structural variant, SV)是指长度≥50 bp的基因组序列变异类型,包括重复、缺失、插入、倒位和易位。相较于广泛研究的单核苷酸变异(Single nucleotide variant, SNV),SV 影响的基因组序列规模更大(约 20 Mb),远超 SNV(约 5 Mb),因此对人类表型多样性和疾病易感性的影响更为显著。近年来,以千人基因组计划、gnomAD 和人类基因组结构变异联盟等为代表的研究,利用短读长(Short-read sequencing, SRS)和长读长测序(Long-read sequencing, LRS)技术,系统描绘了不同人群的 SV 特征,并揭示了其潜在功能。然而,这些研究缺乏对 SV 功能的实验验证,导致SV的基因型与表型之间的因果关系尚不明确,限制了我们对人类进化历史和表型多样性的深入理解。
2025年2月10日,复旦大学进化生物学中心樊少华研究员及金力院士团队、华东师范大学关玉婷研究员团队、美国贝勒医学院Fritz J Sedlazeck副教授团队联合在《Nature Communications》杂志在线发表了题为 “Long-read sequencing of 945 Han individuals identifies structural variants associated with phenotypic diversity and disease susceptibility”的研究。该研究基于 945 名汉族人的长读长全基因组测序数据,共鉴定 111,288 个SV,其中 24.56% 为新发现变异。结合人类表型、多组学数据及小鼠敲除模型,研究发现:1)一个源自现代人类、尼安德特人和丹尼索瓦人共同祖先的 SV位于GSDMD 基因座,可影响骨密度和顺铂诱导的急性肾损伤表型;2)一个现代人类特有的 SV 位于 WWP2 基因座,与身高、体重、脂肪、颅面部表型及免疫相关。此外,研究结果表明,GSDMD 基因座的SV可作为一种快速且经济有效的生物标志物,用于评估顺铂诱导的急性肾损伤风险。
基因组结构变异的研究可追溯至核型分析技术的应用,该技术能够检测 3 Mb及以上的染色体非整倍体,并识别导致多种遗传综合征的染色体畸变1,2。随着细胞遗传学技术(如荧光原位杂交)和分子检测方法(如细菌人工染色体、比较基因组杂交等)的发展,全基因组范围内的 SV 解析成为可能3。这些方法可检测千碱基至兆碱基规模的 SV。SRS的兴起及SV检测算法的开发,极大提升了SV及其断点的解析精度4。千人基因组计划4等大规模研究进一步加深了对 SV 多样性及潜在功能的理解。然而,由于SRS读长较短,其在高度重复序列区域或极端 GC 含量区域内的测序准确性受限5,导致大量SV未被检测到。LRS(包括 Pacific Biosciences(PacBio)和 Oxford Nanopore Technologies(ONT))的引入,使得SV检测更加全面5。例如,一项基于LRS的研究对25个不同人群的 32 例个体进行分析,共鉴定约10万个SV,其中仅29.6%可被相同样本的SRS数据检测到6。
作为中国泛基因组联盟(CPC)的延续工作,研究人员基于945名汉族人的长读长全基因组测序数据,构建了高分辨率的结构变异图谱。在对结构变异进行了多维度的真实性评估后,首先对其数量、频率分布及基因组位置进行了表征,并确定了大量此前未被报道的变异(图1)。
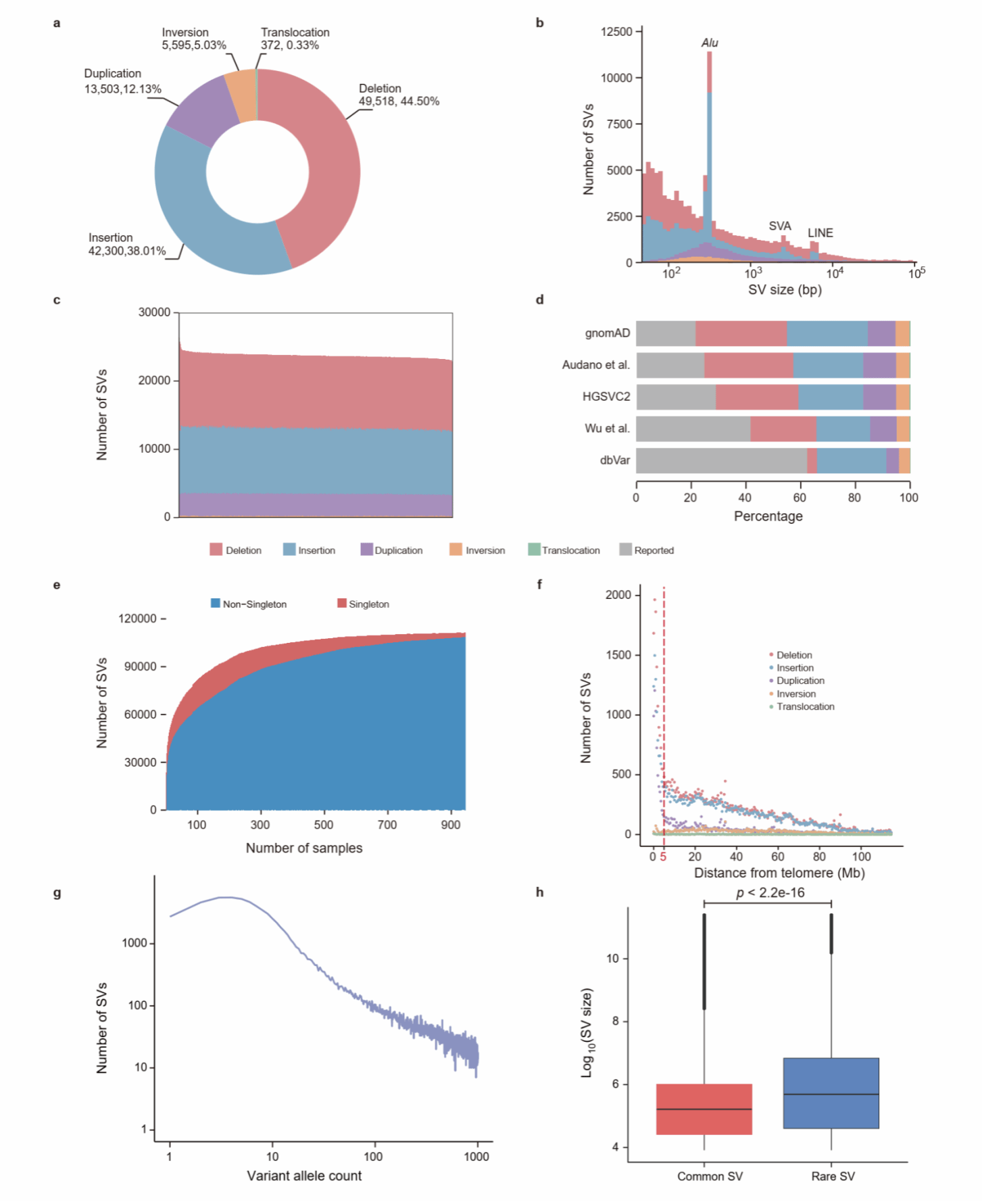
图1. 汉族人群结构变异概况
接下来,为了追溯汉族人群SV的起源,研究者结合黑猩猩基因组、古人类基因组(尼安德特和丹尼索瓦)以及全球现代人类基因组数据进行系统比较,揭示了SV在不同进化阶段的分布模式: 2%的 SV 在人类和黑猩猩基因组中共享,表明其起源可追溯至人-猿分化之前;5% 的 SV 为现代人类、尼安德特人和丹尼索瓦人共同携带,提示这些变异可能起源于三者的共同祖先;32% 的 SV 为现代人类所特有;0.1% 的 SV 由尼安德特人或丹尼索瓦人渗入现代人类基因组,可能与史前时期的基因交流有关;20% 的 SV 为汉族人群特有,其中大多数变异在群体中极为罕见(等位基因频率中位数约 0.003),提示其可能为从头突变(图2)。

图2. 汉族人群结构变异在黑猩猩、古人类和现代人类基因组中的多样性分布
结构变异的功能研究对于理解基因组变异如何影响人类的表型多样性及疾病易感性至关重要。在本研究中,研究人员通过基因注释、GO/KEGG富集分析等方法,在免疫通路富集的基因中,发现一个长度约为2.2 Kb、等位基因频率为0.43的缺失删除了GSDMD最长转录本(NM_001166237.1)的第一个外显子。进一步结合人类表型、多组学数据及小鼠敲除模型,研究者证实该缺失变异影响骨密度和顺铂诱导的急性肾损伤表型(图3)。此外,基于黑猩猩、古人类(尼安德特人和丹尼索瓦人)及千人基因组计划中的现代人类基因组数据,发现该缺失可能起源于现代人类和古人类的共同祖先(图4)。
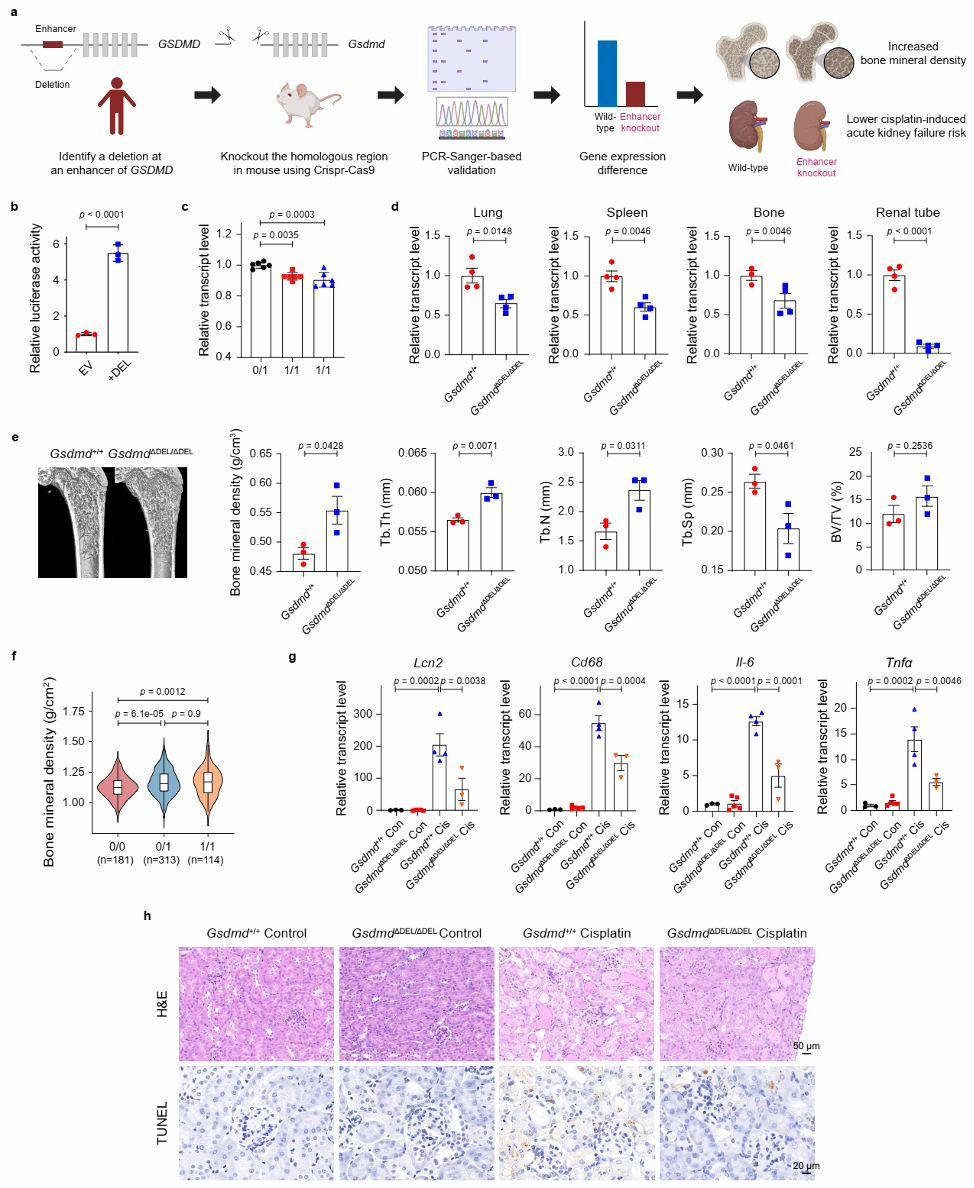
图3. GSDMD基因座的缺失与骨密度和急性肾损伤表型有关
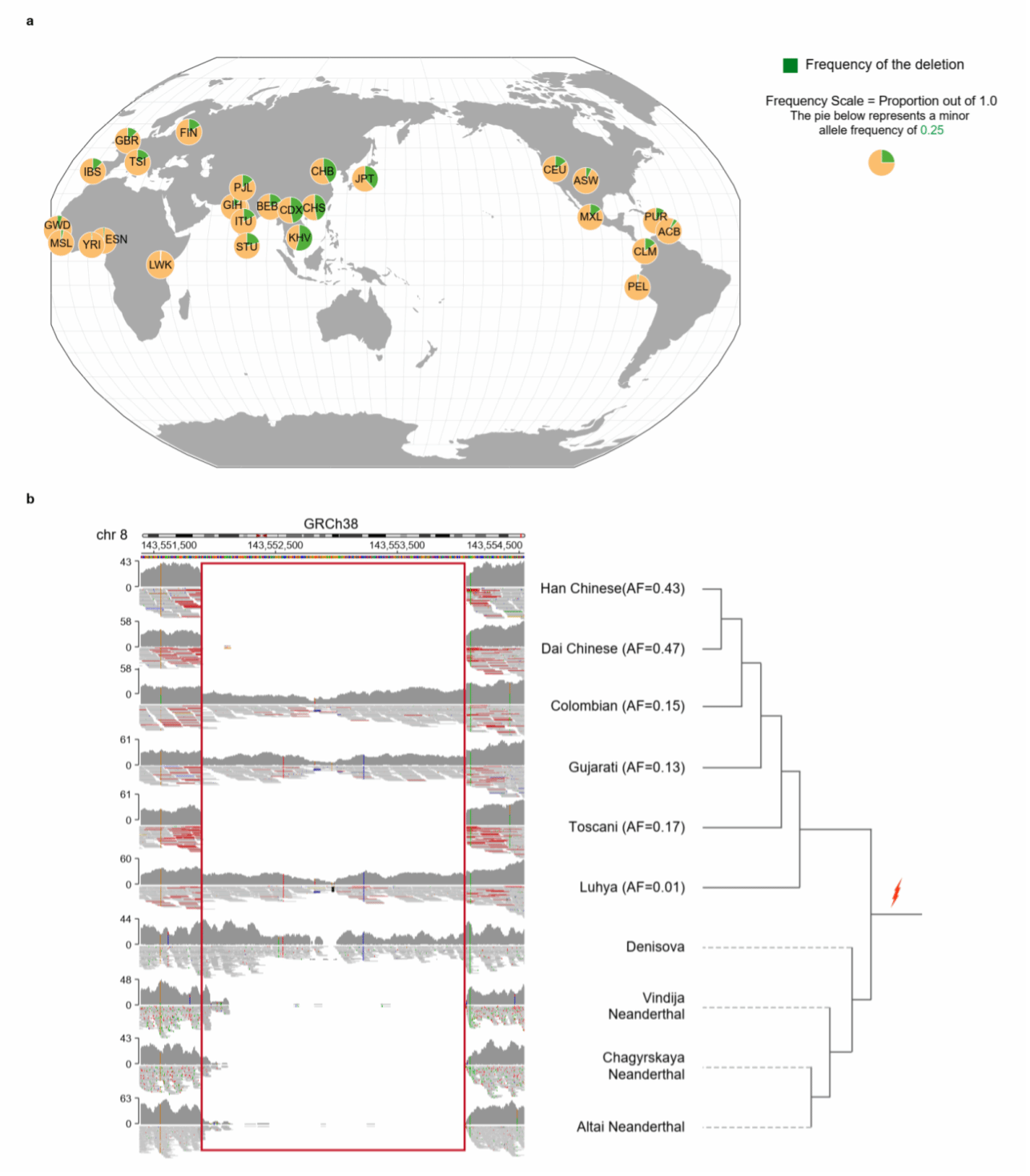
图4. GSDMD基因座的缺失可能起源于现代人类和古人类的共同祖先
由于本研究中95%的结构变异位于非编码区,而这些区域富含功能性调控元件,研究人员进一步探讨了非编码区结构变异的潜在功能。通过基因注释、GO/KEGG 富集分析及小鼠基因敲除表型分类方法,研究者鉴定出一个等位基因频率为0.28 的复杂结构变异,该变异由 229 bp 的插入和 354 bp 的缺失组成,位于WWP2基因(NM_001270454.2)的内含子区。采用与GSDMD基因座SV相似的功能研究策略,研究者证实该结构变异影响人类身高、体脂肪、颅面部多态性及免疫反应(图 5),并且发现该变异可能起源于现代人类的共同祖先(图 6)。

图5. WWP2 基因座的复杂结构变异与身高、体脂肪、颅面部特征和免疫反应相关

图6. WWP2基因座的复杂结构变异可能起源于现代人类的共同祖先
综上所述,研究人员基于长读长测序数据,构建了945名汉族人的结构变异图谱,并对结构变异的来源及基本特征进行了全面表征。通过基因注释、富集分析方法,结合人类表型、多组学数据和小鼠敲除模型,研究者证实了具有不同起源的GSDMD 和WWP2 基因座的结构变异对人类表型多样性和疾病易感性的重要影响。这两个结构变异的表型效应和进化起源结果,帮助我们更深入地理解人类在全球迁徙过程中的基因组多样性和局部适应性。重建这一复杂的历史不仅对理解疾病的地理差异至关重要,同时为设计针对患者的个性化医疗策略提供了重要的理论依据。
复旦大学进化生物学中心樊少华研究员、华东师范大学关玉婷研究员、复旦大学金力院士、美国贝勒医学院Fritz J Sedlazeck副教授为该论文的共同通讯作者,复旦大学博士毕业生龚娇、博士研究生孙蕙茹、华东师范大学硕士研究生汪开元为本论文的共同第一作者。本研究受到国家自然科学基金、国家重点研发计划、上海市科技厅等项目资助。
参考文献
1. Alkan, C., Coe, B. P. & Eichler, E. E. Genome structural variation discovery and genotyping. Nat. Rev. Genet. 12, 363–376 (2011).
2. Weischenfeldt, J., Symmons, O., Spitz, F. & Korbel, J. O. Phenotypic impact of genomic structural variation: insights from and for human disease. Nat. Rev. Genet. 14, 125–138 (2013).
3. Sebat, J. et al. Large-scale copy number polymorphism in the human genome. Science 305, 525–528 (2004).
4. Sudmant, P. H. et al. An integrated map of structural variation in 2,504 human genomes. Nature 526, 75–81 (2015).
5. Sedlazeck, F. J., Lee, H., Darby, C. A. & Schatz, M. C. Piercing the dark matter: bioinformatics of long-range sequencing and mapping. Nat. Rev. Genet. 19, 329–346 (2018).
6. Ebert, P. et al. Haplotype-resolved diverse human genomes and integrated analysis of structural variation. Science 372, (2021).
原文链接:https://www.nature.com/articles/s41467-025-56661-9




